

Research
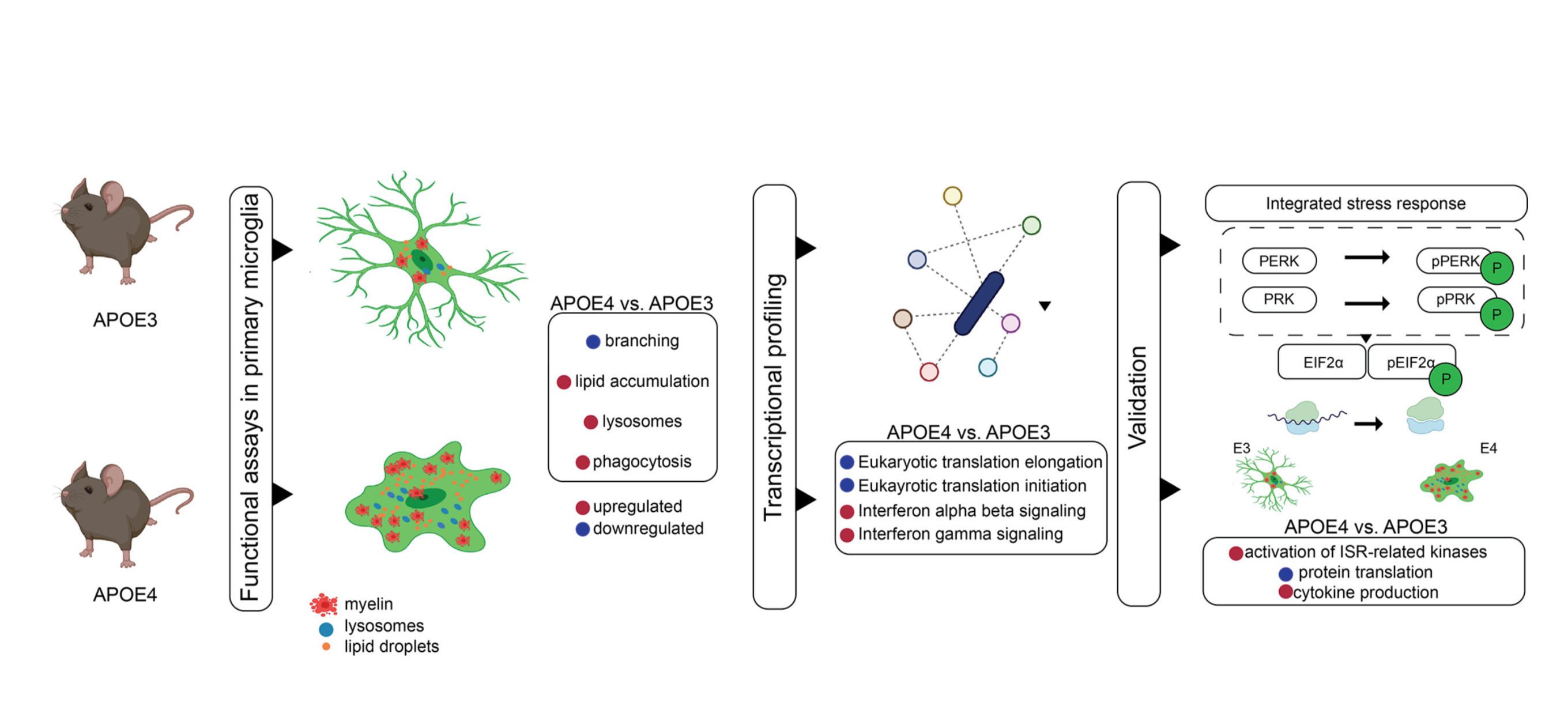
Frontotemporal Dementia (FTD): We use stem cell and genomic approaches to study autosomal dominant causes of tauopathy such as mutations within the MAPT gene. Using crispr genome editing and differentiation of isogenic lines into forebrain organoids we have developed a system that shows age dependent neurodegeneration and recapitulates the selective neuronal vulnerability seen in people with FTD (Bowles et al., 2021:PMID: 34314701). Single cell and bulk RNAseq have identified key pathways that become progressively dysregulated as the organoids age. A second project focused on tauopathies uses an integrative genomics approach to understand the differential risks for sporadic tauopathies associated with the H1/H2 haplotypes. These haplotypes differ from one another by a 1Mb inversion of chromosome 17q21.31. We are using brain tissue and iPSC cultures to determine how this inversion influences chromatin structure and gene expression/regulation to understand how these differences lead to changes in disease risk.
Contact us

Goate Laboratory
Alison M Goate, DPhil
Jean C. & James W. Crystal Professor and Chair
Director, Ronald M. Loeb Center for Alzheimer’s disease
Dept. of Genetics & Genomic Sciences,
Icahn Genomics Institute
Icahn School of Medicine at Mount Sinai
Location
Lab: ICAHN 10-52
Office: ICAHN 10-70C
Phone
Office: 212-659-5672
Email:alison.goate@mssm.edu

Team

Edoardo M Marcora, PhD
Professsor
edoardo.marcora@mssm.edu

Alan E Renton, PhD
Assistant Professor
alan.renton@mssm.edu

Bartek Jablonski
Associate Director
bartek.jablonski@mssm.edu

Ellie Zhang
Executive Assistant
ellie.zhang@mssm.edu

Charlotte Labrie-Cleary
Lab Manager
charlotte.labrie-cleary@mssm.edu

Brian Fulton-Howard, Ph.D
Senior Scientist
brian.fulton-howard@icahn.mssm.edu

Ania Podlesny-Drabiniok, PhD
Instructor
anna.podlesny-drabiniok@mssm.edu

Tulsi Patel, PhD
Instructor
tulsi.patel2@mssm.edu

Carmen Romero-Molina, PhD
Instructor
carmen.romeromolina@mssm.edu

Francesca Garretti, PhD
Postdoctoral Fellow
francesca.garretti@mssm.edu

Chiara Pedicone, PhD
Postdoctoral Fellow
chiara.pedicone@mssm.edu

Hyo Lee, PhD
Postdoctoral Fellow
hyo.lee@mssm.edu

Michael Sewell, PhD
Postdoctoral Fellow
michael.sewell@mssm.edu

Hamilton Oh, PhD
Postdoctoral Fellow
hamilton.oh@mssm.edu

Alexandra E. Münch
Graduate Student
alexandra.munch@icahn.mssm.edu

Grace Peppler
Graduate Student
grace.peppler@icahn.mssm.edu

Nicholas Church
Graduate Student
nicholas.church@icahn.mssm.edu

Jeanne Kim
Graduate Student
jeanne.kim@icahn.mssm.edu

Alexander Frank
Graduate Student
alexander.frank@icahn.mssm.edu

Sun Hao
Graduate Student
hao.sun@icahn.mssm.edu

Danielle Picarello
Associate Computational Scientist
danielle.picarello@mssm.edu

Anthony Walley
Associate Researcher
anthony.walley@mssm.edu

Joshua Orrick
Associate Researcher
joshua.orrick@mssm.edu

Jae-Won Jang MD, PhD
Visiting Scholar
jaewon.jang@mssm.edu

Publications
Goate Lab Gatherings















News



Alumni

Kathryn Bowles, PhD

Kam-Meng Tchou-Wong, PhD

Anna Pimenova, PhD

Shea Andrews, PhD

Julia TCW, PhD

Anastasia Efthymiou, PhD

Saima I. Machlovi, PhD

Manav Kapoor, PhD

Laura-Maria Oja

Franco Abbate, PhD

Gloriia Novikova, PhD

Benjamin Jadow

Rozhan Khaleghi

Riana Khan

Bianca T. Esposito

Jo-Anne Elikann

Sarah Neuner, PhD

Joseph Kakkis

Jessica Schulman

Yiyuan Liu, PhD

Rose Temizer

Travyse Edwards

Iya Prytkova

Nadia Harerimana

Alisha Aristel

Manon Herbinet

Wen Yi See

Marcelina Ryszawiec

Sarah Weitzman