
Research
 Alzheimer disease (AD), the most common cause of dementia in the aged, is a neurodegenerative disorder caused by severe neuronal loss and synaptic abnormalities in the hippocampus and neocortical regions of the brain. Neuropathologically, the disease is defined by brain accumulation of amyloid plaques (APs) and neurofibrillary tangles (NFTs). AD is a serious humanitarian and financial problem as it afflicts a large number of individuals and is very expensive to care for its victims. Most AD cases are classified as sporadic (SAD) because they lack obvious genetic etiologies. A small percent of all cases however, segregates within families (FAD) suggesting genetic etiologies. Most FAD mutations map on three genes encoding the Amyloid Precursor Protein (APP), presenilin1 (PS1) and presenilin2 (PS2). Close to 200 FAD mutations map on the gene encoding PS1 while about 20 FAD mutations are found in APP and 10 in PS2. PSs are functional components of the γ-secretase proteolytic complexes that process many receptor proteins and APP. PS/γ-secretase processing of APP yields Aβ peptides that aggregate to form APs used to define AD. The mechanisms by which FAD mutants promote neuronal death and AD remain unclear. A common theory suggests that AD is caused by Aβ peptides and its derivatives, but inhibitors of Aβ production or anti-Aβ antibodies show no therapeutic value (for recent review see Robakis 2011). Other evidence indicates that FAD mutants affect cellular functions including inhibition of signal transduction and gene expression. Since SAD and FAD have similar clinical and neuropathological phenotypes, understanding how genetic mutations promote neurodegeneration and AD will also help our understanding of SAD.
Alzheimer disease (AD), the most common cause of dementia in the aged, is a neurodegenerative disorder caused by severe neuronal loss and synaptic abnormalities in the hippocampus and neocortical regions of the brain. Neuropathologically, the disease is defined by brain accumulation of amyloid plaques (APs) and neurofibrillary tangles (NFTs). AD is a serious humanitarian and financial problem as it afflicts a large number of individuals and is very expensive to care for its victims. Most AD cases are classified as sporadic (SAD) because they lack obvious genetic etiologies. A small percent of all cases however, segregates within families (FAD) suggesting genetic etiologies. Most FAD mutations map on three genes encoding the Amyloid Precursor Protein (APP), presenilin1 (PS1) and presenilin2 (PS2). Close to 200 FAD mutations map on the gene encoding PS1 while about 20 FAD mutations are found in APP and 10 in PS2. PSs are functional components of the γ-secretase proteolytic complexes that process many receptor proteins and APP. PS/γ-secretase processing of APP yields Aβ peptides that aggregate to form APs used to define AD. The mechanisms by which FAD mutants promote neuronal death and AD remain unclear. A common theory suggests that AD is caused by Aβ peptides and its derivatives, but inhibitors of Aβ production or anti-Aβ antibodies show no therapeutic value (for recent review see Robakis 2011). Other evidence indicates that FAD mutants affect cellular functions including inhibition of signal transduction and gene expression. Since SAD and FAD have similar clinical and neuropathological phenotypes, understanding how genetic mutations promote neurodegeneration and AD will also help our understanding of SAD.
Areas of Research
PS1 and brain vasculature
Alzheimer’s disease (AD) is a highly heterogeneous, multifactorial disorder, whose complex pathogenic etiology leads to neurodegenerative changes and cognitive decline. Recently, the role of the cerebral vasculature in AD has attracted much attention due to the association of vascular disease and its risk factors with cognitive impairment and AD and of the complex functional relationship between components of the neurovascular unit (NVU) in ischemia and neurodegenerative diseases. Current data show that in AD brains there is significant reduction of microvascular density, increased numbers of fragmented vessels, atrophic “string vessels” and changes of vessel diameter pointing towards endothelial dysfunction in AD. Moreover, data from animal models of AD show that vascular pathology, such as the breakdown of the blood brain barrier (BBB), occur prior to the appearance of amyloid plaques suggesting further that primary vascular pathology may be an etiological factor causing neuronal dysfunction and degeneration.
Since in the CNS neurons and endothelial cells (EC), which make up part of the NVU, work in concert to enable proper brain homeostasis and function, any detrimental events occurring within the microvasculature will affect neuronal activity and vice versa.
Interestingly mutations in presenilin 1 (PS1), which give rise to familial AD (FAD), have been shown to result in profound degeneration of brain vasculature as well as atrophy of neuronal dendrites in a mouse model harboring it.
In our laboratory we study the role of PS1 and its mutations found in FAD on brain endothelial cell function and vascular integrity and we aim to identify mechanisms through which such effects are achieved. We apply in vitro angiogenesis assays using primary endothelial cells from mouse brains and in vivo ischemia model to assess the effects of PS1 FAD mutations in the ability of the brain to resist toxic insults. We also identify changes in brain vasculature in various experimental conditions using MRI imaging, confocal microscopy and molecular biology techniques focusing on protein-protein interactions within angiogenic protein complexes in the brain.
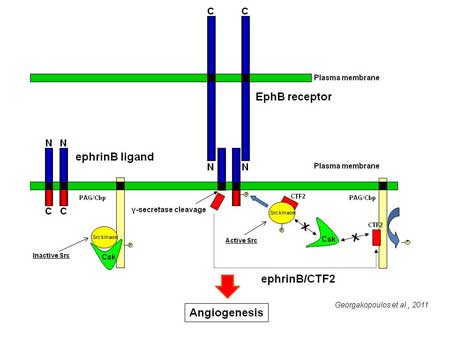
Role of PS and FAD mutants in neoroprotection
To examine the effects of Familial Alzheimer’s disease (FAD) mutations on neurodegeneration, we study the genetics, molecular biology, and biological functions of wild type (WT) and mutant presenilins (PSs) and Amyloid Precursor protein (APP). We found that PS1 FAD mutants cause a loss of γ-secretase cleavage function at ε-sites of substrates manifested by both, decreased production of cytosolic peptides that function in cell signaling and accumulation of γ-secretase substrates. These data support a theory that PS1 FAD mutations promote neurotoxicity by inhibiting γ-secretase-catalyzed ε-cleavage of substrates thus reducing cell signaling while causing accumulation of membrane-bound cytotoxic peptides. We have also found that PS1 mediates brain-derived neurotrophic factor (BDNF)-dependent neuroprotection against toxic insults such as oxidative stress and glutamate toxicity. We currently examine the mechanisms of the PS1-dependent neuroprotection and the effects of FAD mutants on trophic factor-mediated nueroprotection of cortical neurons using both in vitro primary neuronal cultures and in vivo transgenic mouse models.
Role of Progranulin in Frontotemporal lobar degeneration (FTLD)
Recent reports show that progranulin (PGRN) gene null or missense mutations are linked to frontotemporal lobar degeneration (FTLD), a form of dementia characterized by severe neuronal loss in the frontal and temporal brain regions of adult patients. The nature of these mutations suggests that survival of certain neuronal brain populations need expression of both functional alleles of PGRN (haploinsufficiency). We found that PRGN stimulates phosphorylation and activation of neuronal MEK/ERK/p90RSK and PI3K/Akt cell survival pathways and rescues cortical neurons from cell death induced by glutamate or oxidative stresses. Our data showed that extracellular PRGN acts as a neuroptotective factor supporting the hypothesis that in FTLD reduction of PGRN results in decreased survival signaling and neuroprotection against excitotoxicity and other stresses, leading to accelerated neuronal death. We are currently working on the identification of receptors that mediate the cellular effects of PRGN and its mutants involved in FTLD
Contact Us
Robakis Laboratory
Nikolaos K Robakis, PhD
Professor, Psychiatry
Professor, Neuroscience
Location
Lab: Annenberg 22–70, 22–38
Office: Annenberg 22-64
Phone
Office: 212.241.9380
Fax: 212.659.1559
Email
Featured

Publications
2016
Moreno, J.L., et al., Allosteric signaling through an mGlu2 and 5-HT2A heteromeric receptor complex and its potential contribution to schizophrenia. Sci. Signal., 2016. 9(410):
Angeliki M. Nikolakopoulou, Anastasios Georgakopoulosa, Nikolaos K. Robakis. Presenilin1 promotes trypsin-induced neuroprotection via the PAR2/ERK signaling pathway. Effects of presenilin1 FAD mutations. Sciencedirect.com. doi:10.1016/j.neurobiolaging.2016.02.028
2015
Bruban, J., et al., Presenilin 1 is necessary for neuronal, but not glial, EGFR expression and neuroprotection via gamma-secretase-independent transcriptional mechanisms. FASEB J, 2015. 29(9): p. 3702-12.
2014
Roussos, P., et al., A role for noncoding variation in schizophrenia. Cell Rep, 2014. 9(4): p. 1417-29.
Robakis, N.K. and A. Georgakopoulos, Allelic interference: a mechanism for trans-dominant transmission of loss of function in the neurodegeneration of familial Alzheimer’s disease. Neurodegener Dis, 2014. 13(2-3): p. 126-30.
2013
Xuan, Z., et al., Presenilin-1/gamma-secretase controls glutamate release, tyrosine phosphorylation, and surface expression of N-methyl-D-aspartate receptor (NMDAR) subunit GluN2B. J Biol Chem, 2013. 288(42): p. 30495-501.
Barthet, G., et al., Presenilin mediates neuroprotective functions of ephrinB and brain-derived neurotrophic factor and regulates ligand-induced internalization and metabolism of EphB2 and TrkB receptors. Neurobiol Aging, 2013. 34(2): p. 499-510.
2012
Barthet, G., A. Georgakopoulos, and N.K. Robakis, Cellular mechanisms of gamma-secretase substrate selection, processing and toxicity. Prog Neurobiol, 2012. 98(2): p. 166-75.
Meet the Team









Grants
NIH Grants
RO1-NS047229-8 (Robakis, P.I), 07/01/04-06\30\14. NIH/NINDS “PS1 mediates the neuroprotective functions of the ephrinB/EphB system”
2RO1-AG08200-24 (Robakis P.I.). 09\01\1988-08\31\2018. NIH/NIA. “Presenilin1/g-secretase regulate miRNAs and neuronal survival”
5R37 AG017926-12 (Robakis, P.I), 04/01/2001-04/30/15. MERIT Award, NIH/NIA. “PS1 Regulates Processing and Signaling of EphrinB/EphB”
2P50-AG05138-25 (Sano PI), 04/01/87-03/31/15. Alzheimer’s disease Research Center at Mount Sinai School of Medicine “PS1 regulates cleavage and function of ephrinB ligand and EphB receptor.” (Project 3; Dr. Georgakopoulos PI).
Non-NIH Grants
Alzheimer’s Association Investigator Initiated Research Grant IIRG-11-205149 (Anastasios Georgakopoulos, PI) 1/1/12-12/31/14 “Mechanisms of neuroprotection by Presenilin1”
Alzheimer’s Association Investigator Initiated Research Grant IIRG-12-236395 (Junichi Shioi, PI) 12/01/2012-11/30/2015 “Effect of EphB2/CTF2 peptide on NMDAR phosphorylation and signaling”
NARSAD Young Investigator grant, Brain and Behavior Research Foundation (Yonejung Yoon, PI) 2014-2016 “A novel anxiety-associated mechanism of serotonin transporter regulation”